PhenogramViz: Ein Werkzeug zur Spezies-übergreifenden Analyse und Auswertung von Variationen in der Kopienzahl von Genen
Clinical Interpretation of CNVs with cross-species phenotype data.
J Med Genet. 2014 Nov;51(11):766-72. doi: 10.1136/jmedgenet-2014-102633.
PhenogramViz ist ein benutzerfreundliches Werkzeug, das die routinemäßige Diagnostik von Variationen in der Kopienzahl (engl.: copy number variation) eines bestimmten DNA-Abschnittes innerhalb eines Genoms unterstützt, indem phänotypische Information über betroffene Gene visualisiert wird.
Klinische Genetiker sehen häufig eine Vielzahl von CNVs (engl: CNV: copy number variant) innerhalb des Genoms eines Patienten. Zunächst müssen vermutlich krankheitserregende CNVs von vermutlich gutartigen CNVs unterschieden werden. Dies geschieht durch die Untersuchung der Gene, die innerhalb der CNVs liegen. Für Menschen fehlen häufig Informationen über Krankheiten, die durch veränderte Gene hervorgerufen werden. Für Modellorganismen wie Maus oder Zebrafisch werden in entsprechenden Datenbanken phänotypische Folgen abgelegt, die auftreten, wenn bestimmte, zu menschlichen Genen homologe, Gene ausgeschaltet wurden. Der Abgleich von bei Patienten auftretenden Symptomen mit durch Genabschaltung geprägten Erscheinungsbildern (Phänotypen) in den Modellorganismen kann ziemlich aufwändig und Zeit-intensiv werden, insbesondere, da es noch wenig Wissen über die Verbindung von Erscheinungsbildern bei einer Spezies zu Symptomen bei einer anderen Spezies gibt, und entsprechende Softwarewerkzeuge fehlen.
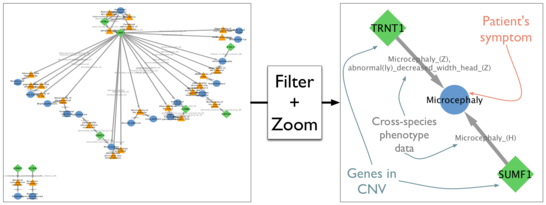
PhenogramViz, eine von uns entwickelte Cytoscape App, soll gerade klinischen Genetikern dabei helfen, lange Listen von CNVs bezüglich der bei einem Patienten aufgetretenen Symptome zu interpretieren. Die App nutzt dabei die interne, Spezies-übergreifende Ontologie Uberpheno. PhenogramViz kann dazu genutzt werden, Beziehungen zwischen Genen und Phänotypen zu visualisieren, indem sogenannte Phenogramme erzeugt werden. Ein sich aus einem Phenogramm ergebener Wert kann dann dazu genutzt werden, bestimmte CNVs zu priorisieren, die wahrscheinlich ursächlich für die Patientensymptome sind.
Dieses Projekt ist eine Kooperation mit der Computational Biology and Bioinformatics des Institut für Medizinische Genetik und Humangenetik der Charité - Universitätsmedizin Berlin