PhenogramViz: A tool for cross-species phenotype analysis and interpretation of copy-number variations
Clinical Interpretation of CNVs with cross-species phenotype data.
J Med Genet. 2014 Nov;51(11):766-72. doi: 10.1136/jmedgenet-2014-102633.
PhenogramViz is a user-friendly Cytoscape App that may help in routine diagnostics of copy number variations by integrating and visualising phenotype information on the affected genes.
Clinical geneticists often are confronted with lists of up to a hundred copy number variants (CNVs) in a patient's genome. Thus, at first, suspected pathogenic CNVs have to be separated from probably benign CNVs, e.g. by investigating the set of affected genes located inside the affected regions. Often, further information on the affected human genes is missing. Thus, model organism databases are consulted, that record the phenotypic consequences of knocking out the homolog of a human gene. However, searching and aligning the model organism's phenotypes with the patient's symptoms can be a laborious and time-consuming task, given the missing integration of phenotype information across species and the lack of dedicated software tools.
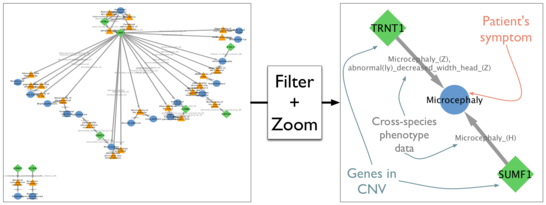
We have implemented a Cytoscape App called PhenogramViz, specifically aimed at assisting the clinical geneticist in interpreting long lists of CNVs, given a set phenotypes seen in a patient. The software tool makes use of the integrated cross-species phenotype ontology Uberpheno. PhenogramViz can easily be used to automatically visualize gene-to-phenotype connections by generating so-called phenograms. We show that a score based on these phenograms can be used to prioritize CNVs that are more likely to be responsible for the patient's phenotype.
This project is a cooperation with the Computational Biology and Bioinformatics Group of the Institute of Medical Genetics and Human Genetics at Charité - Universitaetsmedizin Berlin